Prions: definition, examples, meaning, Structure, Topic
What Are Prions?
Prions are small particles that lack all nucleic acid and are made up of only protein, hence referred to as prions. They are not normal versions of a protein found in the brain – prion protein (PrP) but are misfolded versions of PrP. This misfolding sets off the chain reaction where normal PrP changes to the abnormal prion form to cause scalping and severe neurodegenerative diseases.
NEET 2025: Mock Test Series | Syllabus | High Scoring Topics | PYQs
NEET Important PYQ's Subject wise: Physics | Chemistry | Biology
New: Meet Careers360 B.Tech/NEET Experts in your City | Book your Seat now
- What Are Prions?
- Characteristics Of Prions
- Difference between prions and other infectious agents (bacteria, viruses, fungi).
- Biology Of Prions
- Mechanism Of Disease Progression: Pathogenesis of Prion diseases
- Impact on the Nervous System
- Treatment And Prevention
- Recommended Video on Viruses, Viroids and Prions
The credit for identifying prions goes to Stanley B. Prusiner, who in the 1980s discovered this new pathogen which he came to be associated with the Nobel Prize in Physiology or Medicine in 1997. Prions are since been an important subject of interest in biology and medicine due to their contribution to fatal diseases like CJD and BSE or mad cow disease. Knowledge about prions is necessary for the creation of diagnostic tests and treatments for these diseases and other similar diseases.
Characteristics Of Prions
The mode of action and the conformation of prions make them highly peculiar and categorically different from all other pathogens.
Structure And Composition
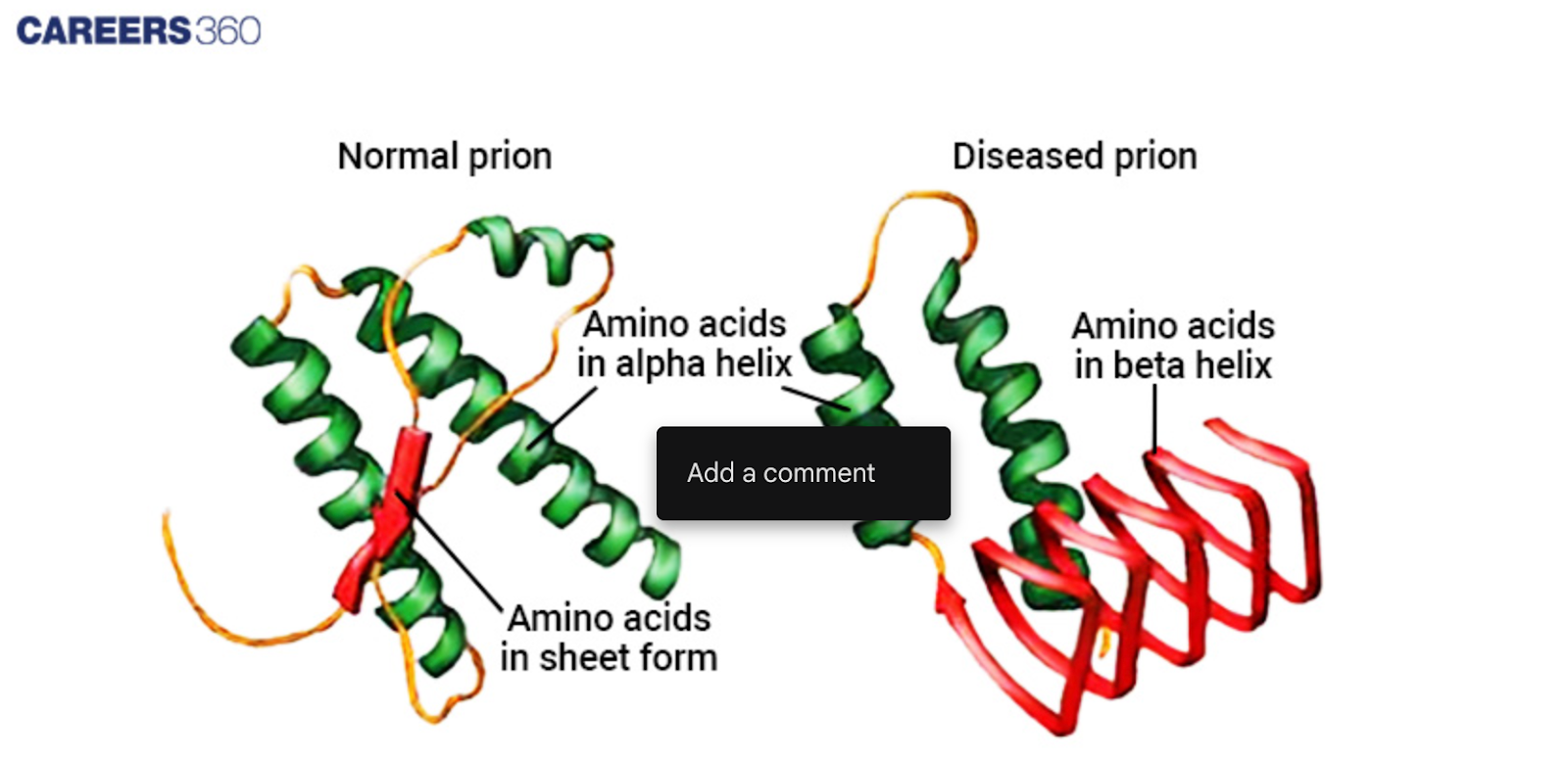
Prions do not have nucleic acids, hence they are not viruses or bacteria but are composed of protein only. The first part is a scrapie prion protein which is an abnormal fold of the cellular prion protein, and conformational isomer.
The specific feature of the misfolded protein is the presence of β-sheets, but not α-helices, as in the case with the normal protein. This structural change is significant because it lets prions alter normal protein into the pathogenic configuration.
Prions are equally very resistant to proteases, these are enzymes that can degrade proteins. This resistance supports their build-up in the environment and dissemination through populations of cells and organisms.
Diagram showing normal and misfolded protein in Prion.
Difference between prions and other infectious agents (bacteria, viruses, fungi).
Feature | Prions | Bacteria | Viruses | Fungi |
Nature | Protein only (misfolded prion protein) | Single-celled microorganisms | Acellular particles with genetic material (DNA or RNA) | Eukaryotic organisms (single-celled or multicellular) |
Genetic Material | None | DNA | DNA or RNA | DNA |
Size | Extremely small | Larger than viruses; typically 0.2-2.0 µm | Smaller than bacteria; typically 20-300 nm | Larger than bacteria; variable sizes |
Cellular Structure | None | Prokaryotic cell structure | No cellular structure (acellular) | Eukaryotic cell structure |
Reproduction | Induces misfolding of normal proteins | Binary fission | Requires host cell machinery to replicate | Spore formation, budding, or binary fission |
Pathogenic Mechanism | Protein misfolding and aggregation | Toxins, invasion, and immune response | Hijacks host cell machinery for replication | Tissue invasion, toxin production |
Examples of Diseases | Creutzfeldt-Jakob disease (CJD), Bovine spongiform encephalopathy (BSE) | Tuberculosis, Streptococcal infections | Influenza, HIV/AIDS, COVID-19 | Athlete's foot, Candidiasis, Ringworm |
Treatment | No known cure; symptomatic relief only | Antibiotics | Antiviral drugs, vaccines | Antifungal drugs |
Resistance to Environmental Factors | Highly resistant to heat, UV radiation, and disinfectants | Variable; some form spores | Generally sensitive to heat and disinfectants | Varies; spores can be highly resistant |
Biology Of Prions
Transmissible spongiform encephalopathies or TSEs are a group of fatal neurological disorders in which prions play roles as the causative agents. These diseases are caused by a build-up of abnormal prion proteins in the brain, which in turn, causes degeneration of the brain and finally, death.
Prion Diseases (Transmissible Spongiform Encephalopathies - TSEs)
TSEs are transmitted through company consumption and impact people and animals. The features which differentiate this group are long incubation periods, spongiform changes in the damaged part of the brain and no immune response to the prion.
Classification
Prion diseases can be classified based on their origin:
Sporadic:
Be unpredictable and have no identifiable traits that could be a warning sign that it could happen.
Creutzfeldt-Jakob disease can be of various subtypes but the most frequent is sporadic Creutzfeld-Jakob disease and it is responsible for 85% of all CJD cases.
Familial:
From a genetic makeup influencing the prion protein gene (PRNP).
Educate diseases for example, Familial Creutzfeldt-Jakob Disease (CJD), Gerstmann-Sträussler-Scheinker Syndrome (GSS), and Fatal Familial Insomnia (FFI).
Make up 10-15% of all prion disease cases.
Acquired:
Consequence of getting infected by prions from food products, medical procedures, or by any other means.
- Some of these are the variant Creutzfeldt-Jakob Disease which results from BSE-tainted food, Kuru from the West Africans’ tradition of cannibalism, and iatrogenic CJD from surgeries.
These cases are however limited but very important particularly because they are infectious.
Examples Of Prion Diseases
Some diseases caused by prions are:
Creutzfeldt-Jakob Disease (CJD)
Description: This disease is in the list of human prion diseases that are characterized by progressive and fatal neurodegeneration.
Types:
Sporadic (sCJD): It is not related to any certain timeline or period.
Familial (fCJD): Inherited.
Iatrogenic: by medical interventions.
Variant (vCJD): Associated with meat products that infected BSE.
Kuru
Description: A tropical neurological disease that was discovered among the fore people of Papua New Guinea.
Transmission: In vain, cut off the arms, leaves in the hands of the dying, the hearts were eaten Aimarams alongside the urine and faeces – throw it into the mud, and the meat is not made.
Symptoms: Shaking, imbalance and components of the neurodegeneration process.
Bovine Spongiform Encephalopathy (BSE or Mad Cow Disease).
Description: Influenced cattle tend to spongy degeneration of the brains.
Transmission: From contaminated feed, the animals take in the feed that has been infected with the bacteria thus causing the disease.
Impact: That can be passed on to humans in the form of vCJD.
Chronic Wasting Disease (CWD)
Description: Impacts deer, elk, and moke species include:
Symptoms: Slimming, transformations in behaviour, and brain degradation.
Transmission: By contact with fluids that come out of our bodies and surfaces that are touched by several people every day.
Mechanism Of Disease Progression: Pathogenesis of Prion diseases
The diseases include Creutzfeldt-Jakob Disease, Gerstmann-Sträussler-Scheinker Syndrome and many others are neurological disorders which are due to the abnormal post-transformation of normal cell surface prion proteins (PrP^C) into abnormal cell surface proteins (PrP^Sc). PrP^Sc is resistant to proteases and preferentially accumulates in an insoluble form, which is the form of insoluble fibrils. These fibrils build up in the brains upsetting the normal running of neurons and causing neurodegeneration.
Impact on the Nervous System
Neuronal Damage: The increase of PrP^Sc results in neuronal degeneration that involves the regions of the brain that control memory, voluntary muscle movements, and balance.
Spongiform Changes: The brain tissue acquires a spongy consistency due to vacuolation due to neuronal cell death and disruption of cell membranes.
Inflammatory Response: Microglia and astrocytes are activated by prions leading to neuroinflammation, but no classic immune response is called for.
Clinical Manifestations
The clinical course of the disease progresses through various stages, each with distinct neurological and psychiatric features.
Symptoms And Signs
Early Symptoms: This may comprise of deficit in memory, confusion, psychiatric disorders such as depression, anxiety, altered sensory experiences or sensations.
Motor Symptoms: Degeneration to ataxia, mere tremors, myoclonic jerks, and subsequent dementia.
Advanced Stages: Alzheimer's dementia, akinetic mutism and terminal stages are coma.
Progression Of Symptoms.
Rapid Onset: Sporadic and variant may be rapidly progressive, hence may lead to death within a few months to a couple of years.
Variable Course: Thus, the onset and the course of pathological changes in familial forms can be different due to the variety of genetic mutations.
End-Stage: Total involvement of the brain and limbs along with the fatal outcome primarily due to the development of complications, such as pneumonia or renal shutdown.
Treatment And Prevention
Methods that can be adapted to prevent the diseases are:
Current Treatment Strategies
Symptomatic Relief: Covers general aspects, signs and so on such as pain and anxiety.
Supportive Care: It offers ‘’Comfort Care’’ to enhance the patient’s quality of life.
Experimental Therapies: Currently there is research that is being done on possible cures in the form of drugs that address themselves to controlling the replication of prion and protein misfolding.
Prevention Strategies
Food Safety: Laws that have been put in place to curb transmission through meat products.
Screening: Screening for blood, and tissues to minimize the iatrogenic transmission hazards.
Decontamination: Finding environments which are appropriate for washing the tools, the accessories and the equipment which are used in the medical field.
Education: A review of prion diseases and their risks to enhance the knowledge level of people.
Recommended Video on Viruses, Viroids and Prions
Frequently Asked Questions (FAQs)
The prions are the pathogen that transmits diseases through a process that leads to the conversion of normal cell prion proteins, PrP^C into an abnormal form known as PrP^Sc. As noted above, these abnormal prions deposit at the synapses of the brain and cause neuronal damage, spongiform changes and consequently neurodegeneration, which is a hallmark of diseases by prions or Transmissible Spongiform Encephalopathies (TSE).
Some of the symptoms of the prion disease may be rapid memory loss or confusion, behavioural changes like anxiety or depression, difficulty in movement, tremors, late-stage dementia, and poor coordination.
Diagnosis of prion diseases is clinical as supported by imaging and laboratory investigations which may include imaging studies such as MRI and lab tests which may detect abnormal prion proteins in the cerebrospinal fluid or biopsy specimens. Subsequently, the definitive diagnosis is reserved, when by histological examination of the brain tissue typical spongiform changes and prion protein deposition are seen in the post-mortem examination.
As of now, there is still no way to eliminate or cure prion diseases, though various medications are being developed to counter them. Management is mainly on manifestations management, patient support and experimental treatment that is oriented at trying to slow down the disease process. Prion diseases do not have a cure and are typically lethal; however, the management focuses mainly on enhancing the patient’s quality of life.
Prevention of prion diseases includes:
Measures to control the spread of the disease through the incorporation of stiff measures that curtail spreading through meat products, especially from infected cattle affected with BSE.
Elimination of blood transfusions and transplantations so that the risk of spreading the disease through transplantation is averted.
Coming up with reasonable procedures to follow to clean medical equipment and areas where prion is likely to be present.
Raising the knowledge level of the public and the healthcare providers concerning prion diseases and their risks.
Prions consist mainly of misfolded proteins, particularly the prion protein (PrP), which can induce normal proteins in the brain to misfold, leading to neurodegenerative diseases. Unlike viruses or bacteria, prions contain no genetic material (DNA or RNA).
Also Read
30 Nov'24 12:23 PM
28 Nov'24 05:34 PM
25 Nov'24 05:18 PM
23 Nov'24 10:02 AM
22 Nov'24 01:59 PM
21 Nov'24 04:58 PM
16 Nov'24 01:58 PM